Millie e seu pai Earl
As leis de Mendel, assim como quaisquer leis na ciência, são maravilhosas porque elas permitem fazer predições. Por exemplo, uma mulher e um homem, ambos portadores de uma mutação recessiva no mesmo gene, e cada uma de suas crianças tem uma chance de 25% de herdar tanto as mutações como as condições de saúde associadas a elas. Bio 101.
Em contraste ao nosso bizarro novo mundo com cenários de ‘fatos alternados’, ‘múltiplas interpretações’ e ‘ambos são verdadeiros’, a ciência é racional e lógica. Se uma observação parece contrariar o dogma, então investigamos e chegamos à verdade. Foi o que aconteceu para Millie e Hannah, cujas histórias ilustram duas maneiras pelas quais parece que a doença genética pode desviar-se das previsões da primeira lei de Mendel: aquela em que os genes separados, uma cópia do pai e uma da mãe, no esperma e nos óvulos, e que se reúnem na fertilização. (Vou comentar a engenharia embrionária no final.)
A situação de Millie é cada vez mais comum: o exoma ou o sequenciamento do genoma do trio – criança, mãe e pai – revela uma nova (“de novo”) mutação dominante na criança, causando uma doença que é genética, mas não herdada.
A situação de Hannah é muito mais rara: herdar uma dose dupla de uma mutação de um dos pais e nenhuma cópia do gene do outro.
A syndrome de Millie e Bainbridge-Ropers
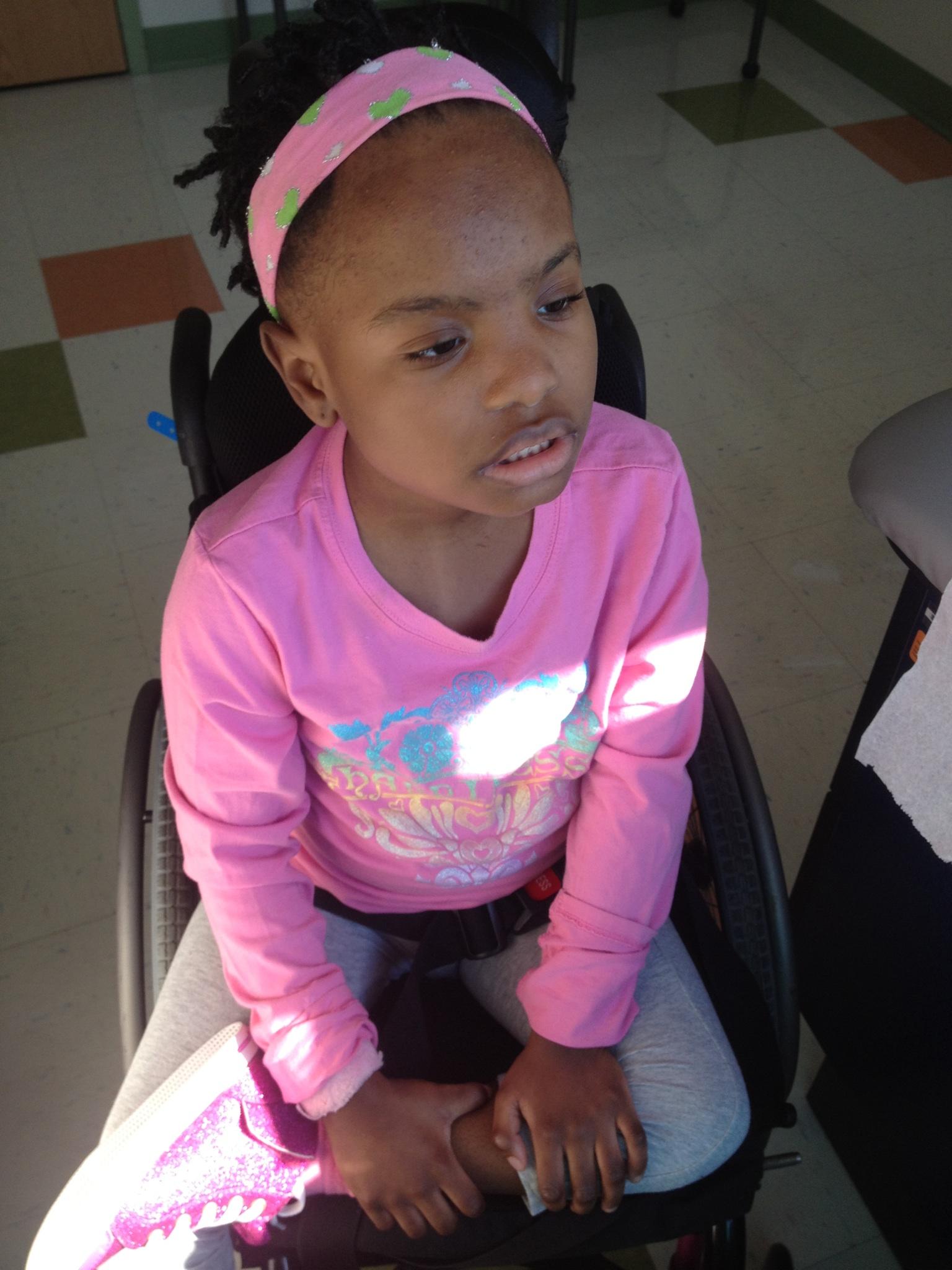
Millie
No primeiro aniversário de Millie, sua cabeça trêmula tornou-se um estranho e constante balançar. Ela não conseguia engatinhar nem sentar, tinha ataques de irritabilidade e vômitos, e mordia as mãos e os dedos.
Em doenças genéticas, hábitos estranhos e certas características faciais podem ser pistas, mas nenhum dos muitos testes, varreduras e biópsias que Millie sofreu levou a um diagnóstico. Nem seus pais eram portadores de quaisquer condições conhecidas que pudessem explicar seus sintomas. Ainda assim, era possível que Millie tivesse uma “apresentação atípica” de uma condição recessiva tão rara que não fosse incluída nos painéis de testes.
Aos seis anos de idade, Millie não conseguia falar, era intelectualmente incapacitada e estava confinada a uma cadeira de rodas, capaz de rastejar apenas alguns metros. Hoje, ela precisa de terapias domésticas intensivas. Mas Millie pode se comunicar. “Ela gosta de olhar para o que a interessa, com um olhar intenso”, disse Angela. Ela adora música country e Beyoncé, e quando algo engraçado acontece e exibe um grande sorriso.
A pediatra de Millie, a Dra. Sarah Soden, sugeriu que o sequenciamento do genoma dela e dos pais, que começou a ser feito na Children’s Mercy Kansas City (onde a criança já recebeu cuidados), pode ajudar a montar as peças clínicas do quebra-cabeça: explicar os sintomas de piora.
Assim, a menina e seus pais, Angela e Earl, tiveram seus genomas sequenciados em dezembro de 2011. A análise dos dados levou meses, mas a equipe do Dr. Soden finalmente encontrou uma mutação genética na criança, mas não em seus pais. No entanto, o gene ASXL3 não tinha sido associado a uma doença infantil. Ainda.
Millie e Dra. Soden
Em uma cópia do gene ASXL3 da Millie estão faltando duas bases de DNA, criando uma cauda inapropriada e encurtando as proteínas codificadas. Desta nova falha surgiram de algum modo os estranhos sintomas. Porque nem Earl nem Angela tem a mutação, a mutação deve ter se originado no esperma ou no óvulo que se tornou Millie.
Desde que o artigo sobre a síndrome de Bainbridge-Ropers foi publicado há três anos, algumas dezenas de indivíduos foram diagnosticados e as famílias formaram um grupo de apoio e uma página no Facebook. Isso é muito bom. Mesmo que uma doença não tenha tratamento, como é o caso da Bainbridge-Ropers, as famílias podem encontrar conforto, depois de uma odisseia de diagnósticos, localizando outras famílias com o mesmo problema. Disse Angela: “Foi um alívio finalmente descobrir o que realmente estava acontecendo com ela, e depois entender que outras famílias sofrem o mesmo problema.”
Hannah e GAN
Hannah Sames vai celebrar seu 13º aniversário e está mostrando o que podem ser sinais precoces de força em seus músculos depois de receber uma terapia genética em sua medula espinhal, no verão passado, para tratar a neuropatia axonal gigante (GAN).
Quando eu conheci a mãe de Hannah, Lori, em 2010, ela me disse que Hannah tinha herdado a mesma mutação de deleção no gene gigaxonina dela e de seu marido, Matt. Naquela época, apenas algumas dezenas de crianças eram conhecidas por ter a doença, e esse número não aumentou muito. Por causa da raridade da doença, eu perguntei se Lori e Matt poderiam ser primos sem saber.

Lori e Hannah Sames (Dr. Wendy Josephs)
A resposta veio apenas a alguns meses atrás: Hannah herdou as suas duas mutações de deleção no gene gigaxonina de Lori, e nenhuma de Matt. Este é um fenômeno muito raro chamado dissomia uniparental (UPD), que significa “dois corpos de um dos pais”. Como Millie, a UPD aparentemente desafia a lei de segregação de Mendel, com um par de cromossomos (ou suas partes) provenientes unicamente de um dos pais, em vez de uma da mãe e uma do pai.
A UPD ocorre durante a meiose, a forma de divisão celular que esculpe óvulos e espermatozoides. E exige dois eventos extremamente raros.
Primeiro, algo dá errado durante a separação de um cromossomo no qual o DNA se replicou para formar duas cromátides, assim como duas linhas onduladas de DNA ligadas no meio. Em vez de essas cromátides se separarem em óvulos diferentes, um par fica preso, resultando em um óvulo com duas cópias do cromossomo 16, que carrega a mutação.
Para uma criança com GAN ter resultado da falha meiótica de Lori, seu óvulo com dose dupla do cromossomo 16 deve ter se encontrado com uma célula de esperma para a qual acabou de faltar o cromossomo 16 – esse é o segundo evento raro. Hannah essencialmente herdou a mutação de sua mãe duas vezes, sem a proteção do cromossomo 16 normal de seu pai.
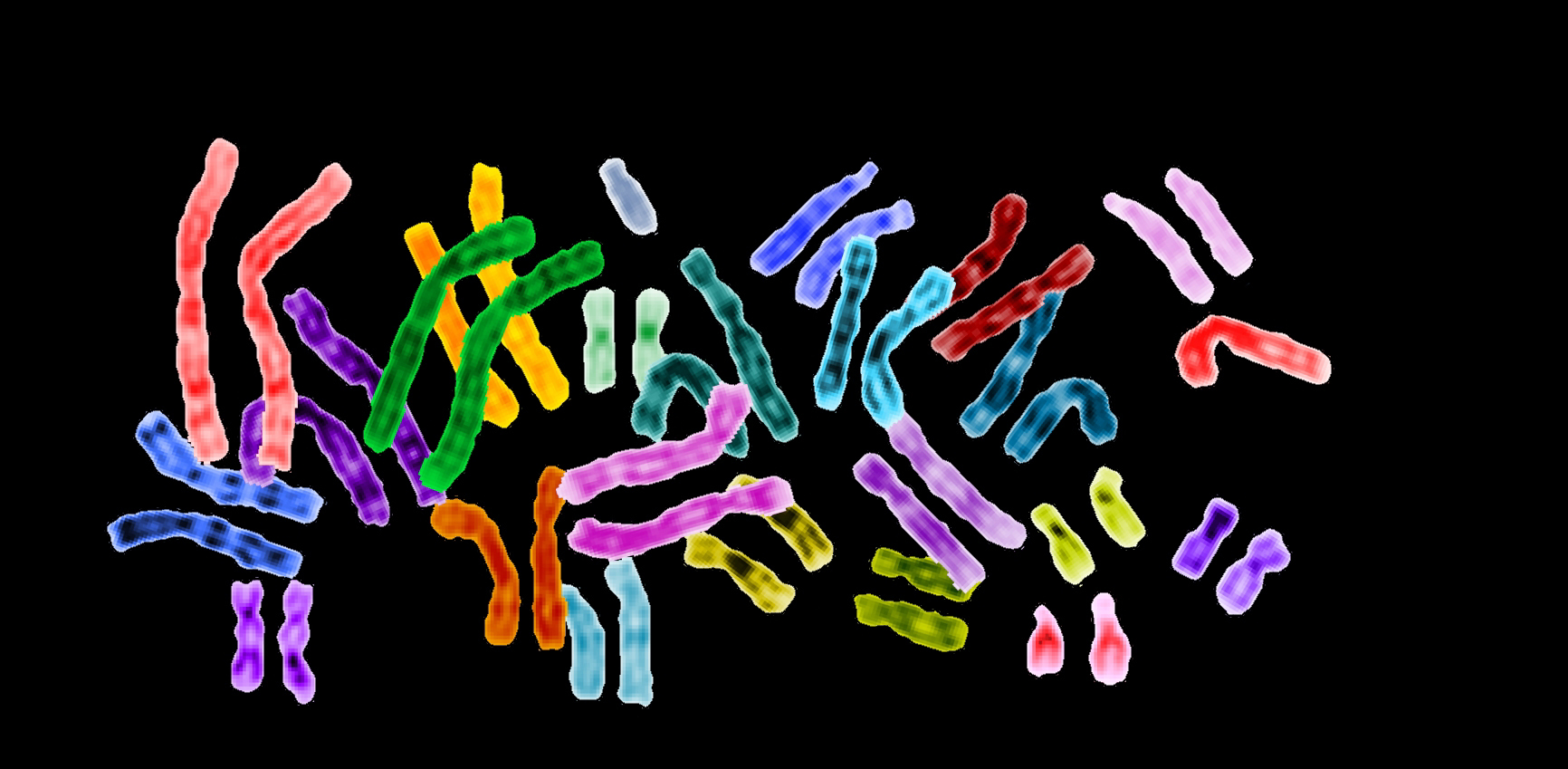
Cromossomos humanos
Esqueça a edição do genoma da linha germinal e ajude as crianças doentes
Enquanto eu escrevia este post, a Academia Nacional de Ciências lançou o tão aguardado tomo sobre o que está sendo chamado, entre outras coisas, “engenharia embrionária”. Em vez de proibir a edição da linha germinativa humana para sempre, o relatório prevê certas situações nas quais a edição de genes ou genomas, usando CRISPR-Cas9, ou alguma outra variação sobre o tema, possa ser implantada para prevenir a doença.
Enquanto eu acho que é ótimo que os raros cenários em que a edição do genoma pode ser útil estão finalmente sendo enunciados, em vez do medo flamejante de que o aprimoramento genético desove bebês projetados, o caso de Millie e Hannah me faz pensar por que não editar o Genoma para prevenir doenças, em primeiro lugar. Para citar o eminente matemático de Jurassic Park, Ian Malcolm,
 “Sim, sim, mas seus cientistas estavam tão preocupados se eles poderiam ou não fazer aquilo, que eles não pararam para pensar se deveriam fazer.”
“Sim, sim, mas seus cientistas estavam tão preocupados se eles poderiam ou não fazer aquilo, que eles não pararam para pensar se deveriam fazer.”Evitar doenças de uma futura criança não é, obviamente, o mesmo que projetar um parque temático, mas assim como a tecnologia do Jurassic Park, eu não posso imaginar porque a edição do genoma em estágios muito precoces de desenvolvimento é necessária.
Mesmo para uma situação familiar extremamente rara em que a transmissão de uma doença hereditária é inevitável, de acordo com as leis de Mendel, existem alternativas, embora não criem uma criança “biológica”: substituir, selecionar ou adotar.
- Uma tecnologia de reprodução assistida pode substituir o esperma (inseminação intrauterina) ou o óvulo (doação de óvulos) que carregam a mutação.
- Um pouco mais além no desenvolvimento, o diagnóstico genético pré-implantacional pode selecionar embriões concebidos in vitro livres da mutação herdada ou espontânea. Ou seja, nós já temos controle de qualidade dos embriões.
- Adoção é uma alternativa óbvia para projetar uma criança.
 Em vez de substituir genes errantes no início do desenvolvimento pré-natal, ou mesmo antes, acho que devemos nos concentrar em ajudar as Millies e as Hannahs que já não são óvulos fertilizados ou embriões precoces, mas são crianças. Esta ajuda já está começando para Hannah, graças à tecnologia de terapia genética que vem sendo desenvolvida desde 1990. A vez de Millie ainda não chegou.
Em vez de substituir genes errantes no início do desenvolvimento pré-natal, ou mesmo antes, acho que devemos nos concentrar em ajudar as Millies e as Hannahs que já não são óvulos fertilizados ou embriões precoces, mas são crianças. Esta ajuda já está começando para Hannah, graças à tecnologia de terapia genética que vem sendo desenvolvida desde 1990. A vez de Millie ainda não chegou.
Então, vamos – sim – definir regras para editar a linha germinal humana – mas vamos também considerar se este tipo de intervenção será mesmo necessário em nosso mundo superlotado.
Explore mais: Mutations in ASXL3 cause problems similar to Bohring-Opitz syndrome
Journal reference: Genome Medicine
Provided by: Public Library of Science
feedback to editors
This story is republished courtesy of PLOS Blogs: blogs.plos.org.
Leia mais: https://medicalxpress.com/news/2017-02-defying-mendelian-genetics-embryo.html#jCp